
Clinical Trial Supply Failures Start Before First Patient In
A conversation with Hedley Rees

Clinical trial supply challenges often appear during study start-up, enrollment, or global trial execution, but many of the underlying causes originate much earlier. Decisions made during nonclinical development and early CMC activities can shape manufacturing flexibility, inventory strategy, forecasting assumptions, and supply chain resilience long before first patient in (FPI). When those decisions are made without considering future clinical supply requirements, sponsors may encounter avoidable constraints later in development, including limited manufacturing responsiveness, inventory shortages, supply chain complexity, and increased operational risk.
As clinical trials become more complex and increasingly dependent on global networks of manufacturers, depots, logistics providers, and technology platforms, the connections between early development choices and downstream supply execution become more important than ever. Understanding where those connections exist can help clinical supply teams identify risks earlier, improve planning assumptions, and build more resilient supply strategies throughout the development life cycle.
In this Q&A, Elizabeth Urbanek, executive editor of Clinical Supply Leader, speaks with Hedley Rees, pharmaceutical supply chain expert and author, about the early decisions that influence clinical trial supply performance, the disconnects that often emerge between development and supply planning, the role of forecasting and RTSM systems, the realities of outsourced supply networks, and the operational risks that become visible only after enrollment begins.
Why do so many clinical trial supply failures originate before FPI, and what are the earliest design choices that lock in that risk?
Despite its crucial importance, nonclinical development is often treated as a box-ticking exercise by the prospective clinical trial sponsor (CTS). There appear to be a few reasons for this:
- It does not generate headline value like clinical milestones.
- It is perceived as a regulatory requirement rather than a strategic lever.
- Timelines are compressed to get to clinic as quickly as possible.
- Responsibility is frequently outsourced with limited integration into overall program strategy.
In practice, of course, the nonclinical supply chain for drug substance (DS) (known as active pharmaceutical ingredient [API] in the EU) is the foundation of future program
success. It is where the most important supply chain decisions must be made. In all too many cases, however, those decisions are delayed until later in the development program, and later, and later again, until the issues begin to emerge and mount up.
Figure 1 depicts the catalog of issues that I have encountered during my years working and consulting in the pharmaceutical supply chain – small molecule and biologics.
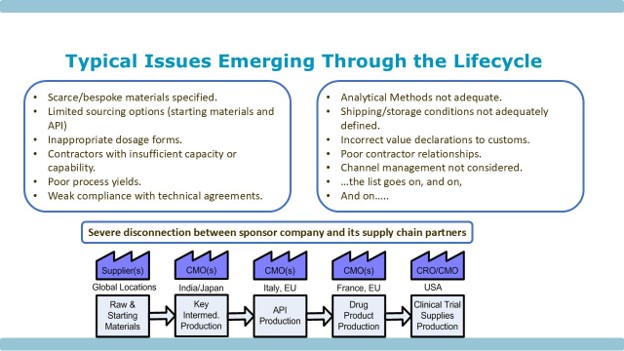
Figure 1: Typical Issues Emerging Through the Development Life Cycle
Every one of the issues identified has the potential to stop a development program in its tracks, if not remediated within a regulatory acceptable timeframe – and those timeframes can be long. In addition, there is the geographic spread of the physical supply chain across the globe, where each and every stage could suffer from the issues above.
In answer to the question “what are the earliest design choices that lock in that risk?”, it seems the absence of supply chain design choices is the real concern.
In many cases, the consequences do not become visible until clinical supply plans are being built, when teams are trying to establish manufacturing timelines, define inventory strategies, and determine whether sufficient material will be available to support study start-up and patient enrollment.
In my professional opinion, that can only be solved by turning drug development on its head, by focusing on the end users of the prospective medical intervention — patients and the clinicians that serve them.
Where do you see the biggest disconnect between protocol design, clinical operations planning, and trial supply execution — and how does that play out once the study starts?
Figure 2 is a schematic representation of the nonclinical supply chain. Typically, a project team will have been formed, with a project manager appointed. The team will consist of CMC and safety staff, and their task is to generate data that will lead to a successful IND application.
In Figure 2, you will see the question “Where is the Supply Chain Thinking?” If I may be so bold – I would suggest that there isn’t any – the focus is purely on generating safety data.
Hence, we see the catalog of issues that emerge further down the line shown in Figure 1.
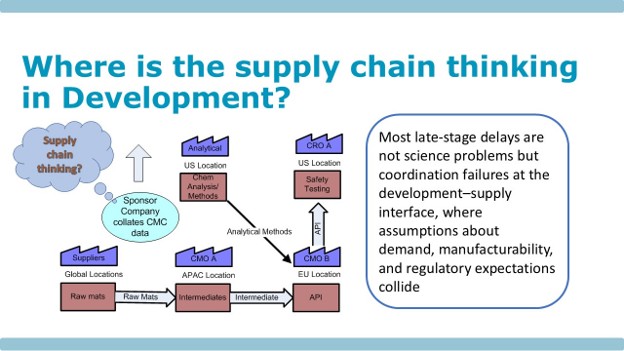
Figure 2: Where Is the Supply Chain Thinking in Development?
This is where the disconnect originates, and it continues into Phase 1 studies where the dosage form is determined as the most convenient to produce, such as soft or hard gel capsules. This has the potential to immediately mask any physicochemical properties of the compound that would prevent it being converted into a more challenging dosage form further on in studies or for market launch.
Then we have scale-up activities moving through Phase 2 a/b and Phase 3, where there is the potential for an unanticipated polymorph to turn what was a safe drug into a toxic cocktail. That has happened to a number of my clients over the years.
In summary, as we see in Figure 2, the disconnect is at the development-supply interface, where the crucial demand and supply planning processes have not been built from the beginning. When the clinical team arrives onboard, establishing trusting relationships with supply teams can be problematic, as can awareness of the vagaries of patient recruitment and other unforecastable outcomes that may occur during clinical operations.
RTSM and forecasting systems are widely relied on to manage trial supply complexity. Where do they genuinely add control, and where do they introduce hidden fragility?
There is an historical perspective to the question that readers should be aware of, if they are not already. In 1990, Michael Hammer wrote an article for the Harvard Business Review, titled Reengineering Work: Don’t Automate, Obliterate. His thesis was that computerization had taken on a life of its own. Existing ways of operating were being computerized without assessments of the value add from these ways of working. In metaphorical terms, organizations were not seeing the wood for the trees. There was a complete disconnection of the needs of an organization from the activities that were being carried out in the name of progress. Hammer advocated radical tree surgery.

Taking the lead from Hammer’s work, and referring to question 1’s answers, the problem we have is the complexity of the multilayered, geographically spread, end-to-end (E2E) supply chain. Computerization will not solve the issues.
RTSM and forecasting platforms can provide tremendous control when the underlying assumptions are sound, allowing teams to plan inventory, manage randomization, and respond to changing enrollment patterns across multiple sites and countries. However, when those assumptions are incorrect or incomplete, the systems can create a false sense of precision, generating supply signals that appear robust but are actually detached from operational reality.
The issues we have discussed above need to be resolved first, then planning and inventory management for clinical trial supplies become an order of magnitude more straightforward.
At what point does the typical outsourced model — spanning CDMOs, depots, packaging, and logistics — shift from enabling flexibility to creating coordination risk?
This is an excellent question, which can be addressed with another historical perspective, this time a personal one. I joined the industry in 1979, working in the U.K. for a U.S. based company, Miles Laboratories, which was subsequently acquired by Bayer the following year.
The production and distribution processes were fully integrated, from the point where raw materials arrived at the goods receiving bay and the finished product was produced within the plant and sent directly to U.K. customers — hospitals, pharmacies, and sometimes even patients at home.
For export markets, the finished products were sent to other Bayer entities around the globe. Those Bayer entities had local presence and distribution capabilities in their own home markets. Links with customers were direct, and the staff at Bayer, the company holding the license to sell the products, could handle customer complaints.
The overriding benefit was a single quality management system (QMS), rather than the multitude of interfacing QMSs in today’s outsourced supply chain arrangements. Information and planning systems covered the entire supply chain, and determining forward and reverse traceability was relatively simple, in stark contrast to today’s lack of transparency in the supply chain.
In clinical development, this complexity often remains manageable until multiple partners must respond to changes in enrollment, demand forecasts, or manufacturing timelines. As each additional organization introduces its own systems, priorities, and decision-making processes, the challenge becomes less about individual performance and more about maintaining visibility and alignment across the network.
With Novo Holdings recently acquiring the CDMO Catalent, I wonder if there will be more industry initiatives to move toward increasingly integrated business models?
How do early nonclinical and CMC decisions translate into constraints on manufacturing responsiveness and inventory strategy once trials are underway?
Nonclinical and early-stage CMC decisions define:
- what the drug is (its molecular and biological behavior)
- how it will be manufactured (process feasibility and scalability)
- what risks are inherent (toxicology, biodistribution, immunogenicity)
- what regulators will expect going forward.
This covers the product’s quality attributes, analytical approach, process knowledge, and scale-up path; those choices shape later manufacturing flexibility rather than merely supporting it. In practice, the more provisional or under-characterized the early package is, the more later changes require comparability work, extra testing, or regulatory interaction.
In terms of manufacturing, if you lock in a process, raw material set, formulation, container closure, or analytical control strategy too early, you usually narrow your ability to respond quickly to demand spikes, supply interruptions, or tech transfer needs. That is especially visible in biologics and cell/gene therapy, where later manufacturing changes can demand substantial comparability evidence and may even create clinical hold risk if not justified well.
Early CMC and nonclinical choices also drive inventory strategy because they determine batch size, expiry profile, allowable overage, lead times, and how much safety stock you need to buffer uncertainty. Clinical supply teams therefore often have to plan inventory around forecast uncertainty, batch failure risk, packaging and labeling lead times, and whether the process can replenish material fast enough to avoid stockouts.
What are the most persistent clinical trial supply risks that only become visible after enrollment begins, despite being predictable in hindsight?
The most persistent supply risks that “suddenly” surface only after FPI are almost always demand‑side issues (actual recruitment, behavior and protocol execution at sites) colliding with brittle, oversimplified supply and forecasting assumptions that were set at start‑up. In hindsight they’re predictable because they stem from structural features of the protocol, the network of sites, and the product itself, but they only truly manifest once real patients, real visit patterns, and real site practices interact with the supply design.
In my work, I have adopted a scenario modelling approach for product launch planning:
- Base case — most likely launch date, most likely volume
- Optimistic case — earliest launch date, highest volume
- Pessimistic case — latest launch date, lowest volume
The next step is to test out each demand load on the supply chain, using a master production schedule, to pressure test the current state supply chain.
It involves exploring the supply chain under the different demand profiles above. The boundary conditions are:
- early launch/high volume
- late launch/low volume.
Inventory strategy (such as safety stock holding) can then be determined based on the modelling. This is described in more detail in Transforming the Pharmaceutical Supply Chain.
About The Expert:
 Hedley Rees is managing consultant at PharmaFlow Ltd., a specialist consultancy challenging the status quo in pharmaceutical supply and manufacturing.
Hedley Rees is managing consultant at PharmaFlow Ltd., a specialist consultancy challenging the status quo in pharmaceutical supply and manufacturing.
Hedley partners with pharma and biotech leaders, investors, and policymakers who recognize that today’s fragmented supply chains, outsourcing dependencies, and opaque practices are no longer fit for purpose. Working across nonclinical, clinical, and commercial stages, Hedley helps organizations redesign end‑to‑end supply networks, rethink CDMO strategies, and build operational models that are resilient, data‑driven, and accountable to patients and society.
His work shines a light on hidden risks, perverse incentives, and gaps between regulatory intent and industry behavior. As an outspoken commentator and advocate for change, Hedley contributes to public and boardroom debates on drug shortages, reshoring, vertical integration, and regulatory reform.
He is the author of Transforming the Pharmaceutical Supply Chain (Wiley, 2025) and Supply Chain Management in the Drug Industry: Delivering Patient Value for Pharmaceuticals and Biologics (Wiley, 2011).