
2024 Trends In FDA Observations For Sterile Drug Manufacturers
By Marzena Ingram, Eliquent Life Sciences; Ajay Pazhayattil, Ph.D., cGMPWorld; Prasanna Sagar, Ph.D., ITAAN Pharma; Subrata Chakraborty, Ph.D., INOPHAR Consulting

Back in 2023, the most common U.S. FDA audit observations for sterile drug product manufacturers highlighted issues in sterile facility design, contamination control, equipment cleaning/maintenance, environmental monitoring, and comprehensive process validation.1 The top CFR sections (Table 1) violated by the inspected facilities in 2023 were sections 211.42, 211.113, and 211.67. Section 211.42, design and construction features, led the list, emphasizing the need for adequate facility design and construction features to uphold the integrity of sterile manufacturing. The second most frequent observation was for section 211.113, control of microbiological contamination, which highlighted the need to control viable microbiological contaminants adequately. Finally, the third most frequent observation was related to section 211.67, equipment cleaning and maintenance, which is associated with observations on the lack of adequate equipment cleaning and preventive maintenance. In this article, we analyze the 2024 observation trends based on Form 483s published by the FDA.2
In 2024, CFR sections 211.113, 211.42, 211.192, 211.100, 211.67, 211.160(b), and 211.22(a)(d) took center stage by collectively contributing to almost 70% of the total observations handed over to sterile facilities (Figure 1). The top observations pertaining to sections 211.113 and 211.42 were consistent with the 2023 trend. However, the second most frequently observed CFR section, 211.192, was for the lack of production record review. The third most frequently observed was shared between sections 211.100, written procedures [and] deviations; 211.67, equipment cleaning and maintenance; and 211.160(b), general requirements, laboratory controls. Section 211.22(a) (d), responsibilities of the quality control unit, rounded out the collective 70% of observations with higher observation volumes.
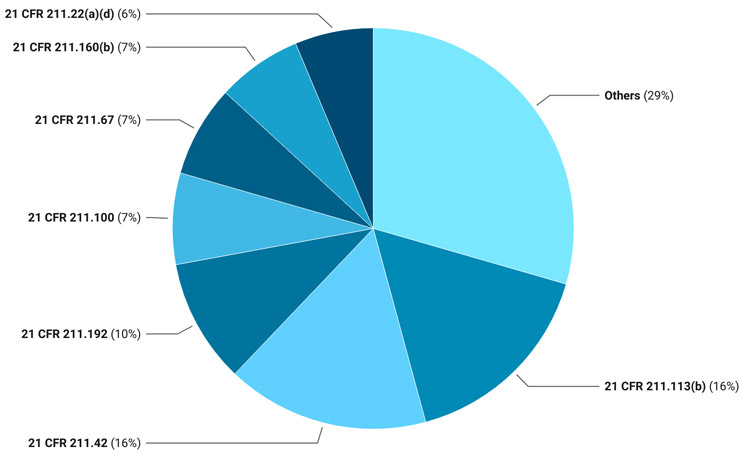
Figure 1: 2024 Pie Chart
21 CFR 211 Sections
- 211.100: Written procedures; deviations
- 211.113(b): Control of microbiological contamination
- 211.160(b): General requirements: laboratory controls
- 211.192: Production record review
- 211.22(a)(d): Responsibilities of quality control unit
- 211.42: Design and construction features
- 211.67: Equipment cleaning and maintenance
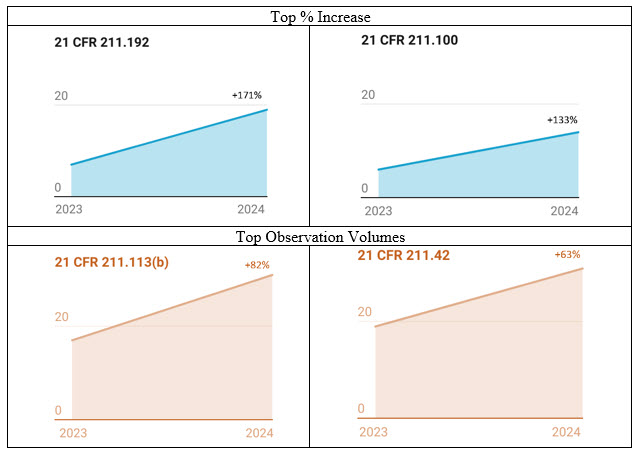
Figure 2: Observation % Increase 2023–24 and Volumes. The numbers are based on the published 483 observations.
Observations With Highest Percentage Growth From 2023 To 2024
CFR section 211.192 saw the highest percentage increase in observations for 2024, a 171% increase (Figure 2). This section highlights the importance of conducting investigations, including cross-contamination investigations. In a media fill failure investigation-related warning letter, the FDA noted that the firm did not adequately consider the operators who performed the cleaning as the likely source of the contamination. Further, the regulator ascertained that the firm failed to ensure appropriate interventions and vial clearance for glass breakage during manufacturing.3 In another warning letter example, the FDA highlighted that the firm had two sterility failures during finished product testing where it identified the contaminating organisms; however, it failed to investigate to determine the root cause of the failures.4 The observations handed over to major injectable manufacturers highlight the deterioration of their investigation robustness. It is imperative that an investigation must be conducted by a technically qualified team with access to the appropriate investigation tools and methodologies.5 The first step is for sterile manufacturing organizations to have high-quality investigator training and a robust qualification program, along with a component of independent external subject matter expert authentication of internal capability.
The second highest percentage increase in observations for 2024 was for section 211.100, showing a 133% increase. Most of the observations in section 211.100 were related to process validation, particularly the insufficient generation of supporting data to evaluate variability. This highlights that the regulator also has acknowledged the importance of addressing variability in sterile injectables processes, expanding the focus beyond the existing emphasis on variability in solid oral site inspections. There have been several instances in which inadequate responses to observations resulted in warning letters. In one case, the FDA identified in the warning letter that the firm lacked supporting data to assess the manufacturing processes' variability (intra- and inter-batch variation). Based on the violation, the regulator highlighted the need for a consultant.6 Organizations that have proactively implemented robust identification of critical process parameters (CPPs) and statistically sound methods to determine the extent of CCPs’ impact on CQAs (all during PV Stage 1 studies) and that have monitoring methods to detect variability during commercial operations (Stage 3) stand to reduce this particular regulatory risk significantly.
Observations With Highest Number Of Citations In 2024
In 2024’s observation outcomes, it was evident that there was a high volume of 483 observations for sterile injectable facilities inspected by the FDA as compared to volumes observed in 2023. Observations related to sections 211.113, control of microbiological contamination, and 211.42, sterile facility design and construction features, continued to garner the most scrutiny from the regulator. In one case related to section 211.113 that resulted in a warning letter, although the organization committed to monitoring interventions to minimize microbiological contamination, the FDA determined that it fell short in its commitment, as the site did not address how it would ensure the aseptic operators would adequately follow the aseptic procedures. The agency highlighted the poor aseptic practices, the inadequacy of gowning, the media fill program, airflow visualization studies, and sterilization qualification.7 This confirms the already observed trend8 of the regulator promoting automation and isolator technology for patient safety. The high volume of aseptic behavioral observation necessitates organizations utilizing RABS technology for aseptic processing to think differently. Adopting industry-recognized training programs9 and implementing practically effective training tools such as virtual reality (VR)10 are essential. VR can infuse training integrity as it eliminates the possibility of introducing human decision-making biases when evaluating aseptic skills of the operator performing cleanroom cleaning, setup, and operations and the microbiologists performing sampling and sterility testing of the product.
Observations related to CFR 211.42, sterile facility design and construction features, were equally high in 2024. The majority pointed toward design elements and the adequacy of environmental monitoring (EM) programs. In a warning letter,4 because of the observations associated with section 211.42, the FDA pointed out that non-viable particulate monitoring, personnel monitoring, active/passive viable air sampling, and sampling of critical surfaces for microorganisms in the aseptic processing area in association with each production batch was not performed. The same warning letter highlighted that the organization did not validate the process for cleaning and disinfecting the cleanrooms where aseptic products were manufactured. There were observation trends for 2024 that were related to the questionable integrity of aseptic operations. In one such case, the FDA noted that even though the firm confirmed that the activities (equipment cleaning, disinfection, and sterilization) would be performed under supervision, including a video review, its response was inadequate as it did not address how the video retention period would be sufficient for QA to review video footage as part of the batch release. Inadequacies in cleaning validation and the need for improvements in the cleaning validation programs also showed up, with special emphasis on the identification and evaluation of all worst-cases elements covering drugs with higher toxicities, high potencies, and lower solubility in their cleaning solvents, characteristics that make it difficult to clean, justified swabbing locations, and establishment of maximum hold times. The FDA determined that the firms needed subject matter support to address these observations.
2023 And 2024 Product Recall Trends And Analysis
Our assessment of 2023 and 2024 recalls (Figure 3) revealed that a large percentage of the sterile drug product (vial) recalls in the United States were associated with:11
- the presence of particulate matter in vials, predominantly glass particles
- CQA failures such as pH, impurity, and assay
- lack of sterility assurance, such as media fill failure and sterility testing failure
- GMP deviations mainly associated with storage/transportation temperature excursions
- labelling compliance-related recalls.
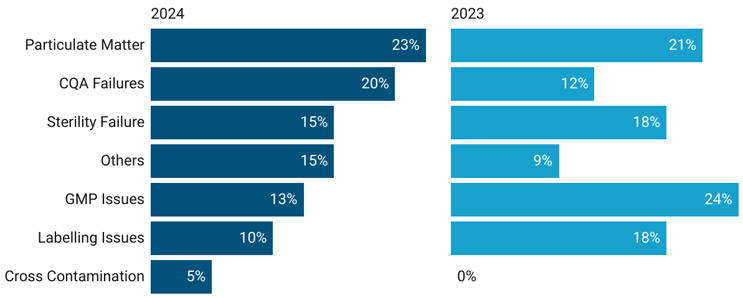
Figure 3: Product Recall (Vials) Trend 2023–24
The continuation of visible glass particles or glass breakage-related recall trends is not encouraging despite the regulator's efforts to resolve the long-standing issue. Where appropriate and possible, especially for terminally sterilized products, the regulator must continue supporting the seamless adoption of alternate packaging technologies like polypropylene vials in the industry.12,13 The availability of polypropylene vials in pre-gamma sterilized packs eliminates the additional handling and extended exposure (vial washing and sterilization) during fill/finish operations that otherwise can result in contamination. Another allowable switch to improve patient and healthcare worker compliance, as well as handling safety, is the switch from sterile bags to polypropylene vials. In addition, it is essential to note that 2024 saw recalls related to cross-contamination. Although the contamination control strategy (CCS) is an Annex 1 requirement, it is also very much relevant to FDA-regulated facilities. Facilities need to revisit their CCS by performing a deep-rooted risk assessment with the support of industry experts to determine the failure modes and risks to patients. This primary action and subsequent remediation (where needed) are necessary to minimize potential recall scenarios and to reduce regulatory inspection risks associated with contamination control.
Conclusion
Analyzing the 483 observations and recalls for 2024 yields interesting insights into the FDA’s current expectations regarding sterile facilities and perhaps foreshadows what is to come in 2025. Foolproof contamination control strategies, which encompass the availability of an integrity verifiable EM program, aseptic monitoring, and robust contamination control risk assessments that justify the existing processes and designs, are all must-haves for aseptic operations, primarily due to their direct implications on patient safety. Manual batch record documentation alone does not stand regulatory scrutiny anymore. The regulator is pressing to adopt the latest digital and manufacturing technologies to secure high-patient-risk products from viable and non-viable contamination. The regulatory and industry efforts indeed are expected to culminate in long-term supply security of sterile drug products for the U.S. market.
References
- Pazhayattil, A.B, Ingram, M. (2024). Safeguarding Sterility: Crucial Insights from 2023 Pharmaceutical Facility Audits, Pharma Focus Europe.
- FDA Data Dashboard: Compliance Dashboards, Inspections, U.S. FDA: https://datadashboard.fda.gov/ora/index.htm
- U.S. FDA, Warning Letter, August 15, 2024: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/eugia-pharma-specialities-limited-681905-08152024
- U.S. FDA, Warning Letter, November 1, 2024: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/frontier-biologics-llc-686059-11012024
- Pazhayattil, A.B, Sharma, S. (2025). Pharmaceutical Manufacturing Deviation and Failure Investigations: Principles, Practices, and Case Studies, AAPS-Springer.
- U.S. FDA, Warning Letter, August 29, 2024: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/zydus-lifesciences-limited-685224-08292024
- U.S. FDA, Warning Letter, July 11, 2024: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/brassica-pharma-pvt-ltd-679005-07112024
- Pazhayattil, A, (2024). A New Year Call for Action: Analyzing 2023 FDA form 483 Observations for Indian Sterile Manufacturing Facilities, Express Pharma.
- PDA, PDA Training and Research Institute: https://www.pda.org/training/pda-training-and-research-institute
- Chakraborty, S. (2024). Conventional Training Vs Virtual Reality (VR) Training, Pharma Machines & Technology.
- U.S. FDA, Recalls, Market Withdrawals, & Safety Alerts: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts
- Sagar, P. Pazhayattil, A.B. (2023). Polypropylene (PP) Vials: An Efficient Primary Packaging Solution for Injectable Drug Products, AAPS Newsmagazine.
- Pazhayattil, A., Sagar, P., Joseph, P. (2023). Time to Start Using Polypropylene Vials in the Industry: A Comprehensive Analysis of Benefits and Applications on Polypropylene Vials, PDA Letter.
About The Authors:
 Marzena Ingram is an independent senior pharmaceutical consultant with extensive QA, technical operations, and process validation expertise. Her specialty is in navigating FDA warning letter scenarios and providing clients with regulatory-compliant solutions. She is known for strategic team leadership. She has driven PV compliance initiatives that have met global regulatory standards and has been involved in establishing industry benchmarks. Additionally, Ingram has built and led specialized teams in high-volume manufacturing settings, ensuring process validation excellence and performance. Ingram has served as VP of ISPE Canada.
Marzena Ingram is an independent senior pharmaceutical consultant with extensive QA, technical operations, and process validation expertise. Her specialty is in navigating FDA warning letter scenarios and providing clients with regulatory-compliant solutions. She is known for strategic team leadership. She has driven PV compliance initiatives that have met global regulatory standards and has been involved in establishing industry benchmarks. Additionally, Ingram has built and led specialized teams in high-volume manufacturing settings, ensuring process validation excellence and performance. Ingram has served as VP of ISPE Canada.
 Ajay Pazhayattil, Ph.D., is an accomplished management consultant and industrial pharmacist with extensive experience spanning solid oral, sterile, and API sectors. He is the founder of cGMP World. Pazhayattil has held key leadership positions with prominent North American brands, generic manufacturers, and CDMOs. His roles include vice president of scientific and regulatory affairs at Capcium, quality director at Eurofins, and associate director at Apotex. He plays a pivotal role in guiding organizations through remediation efforts related to U.S. FDA 483 observations and warning letters. He has been a lead author and contributor to guidance documents published by industry organizations such as AAPS, PDA, ISPE, and RAPS.
Ajay Pazhayattil, Ph.D., is an accomplished management consultant and industrial pharmacist with extensive experience spanning solid oral, sterile, and API sectors. He is the founder of cGMP World. Pazhayattil has held key leadership positions with prominent North American brands, generic manufacturers, and CDMOs. His roles include vice president of scientific and regulatory affairs at Capcium, quality director at Eurofins, and associate director at Apotex. He plays a pivotal role in guiding organizations through remediation efforts related to U.S. FDA 483 observations and warning letters. He has been a lead author and contributor to guidance documents published by industry organizations such as AAPS, PDA, ISPE, and RAPS.
 Prasanna Sagar, Ph.D., is an authority in injectable formulations, bringing over 25 years of extensive experience in injectable R&D. His professional expertise spans the spectrum of formulation design and commercialization of sterile injectable products. A recognized specialist in legal and technical aspects of Paragraph-IV filings, Sagar has successfully contributed to multiple high-stakes ANDA submissions for the U.S. market, ensuring compliance with stringent regulatory frameworks. Sagar is the assignee on several patent applications, and he is an active contributor to the academic and research ecosystem, serving on prestigious academic councils, syllabus boards, and research committees. He holds an M. Pharm and Ph.D. degree in the field.
Prasanna Sagar, Ph.D., is an authority in injectable formulations, bringing over 25 years of extensive experience in injectable R&D. His professional expertise spans the spectrum of formulation design and commercialization of sterile injectable products. A recognized specialist in legal and technical aspects of Paragraph-IV filings, Sagar has successfully contributed to multiple high-stakes ANDA submissions for the U.S. market, ensuring compliance with stringent regulatory frameworks. Sagar is the assignee on several patent applications, and he is an active contributor to the academic and research ecosystem, serving on prestigious academic councils, syllabus boards, and research committees. He holds an M. Pharm and Ph.D. degree in the field.
 Subrata Chakraborty, Ph.D. is the principal advisor and CEO of INOPHAR Consulting and Training. With 27+ years in manufacturing, quality operations, validation, and training, he’s an expert in aseptic processing and contamination control. In his career, Chakraborty spent more than a decade in leadership roles at Pfizer, Novartis, Fresenius-Kabi, and Cipla. He consistently focuses his efforts on advancing pharmaceutical education and industry standards and actively contributes as an author, speaker, and technical reviewer in global industry forums like PDA, ISPE, and SfSAP.
Subrata Chakraborty, Ph.D. is the principal advisor and CEO of INOPHAR Consulting and Training. With 27+ years in manufacturing, quality operations, validation, and training, he’s an expert in aseptic processing and contamination control. In his career, Chakraborty spent more than a decade in leadership roles at Pfizer, Novartis, Fresenius-Kabi, and Cipla. He consistently focuses his efforts on advancing pharmaceutical education and industry standards and actively contributes as an author, speaker, and technical reviewer in global industry forums like PDA, ISPE, and SfSAP.