
Comparability Considerations For mRNA Product Development
By Elaine Peters, Genentech

Following the success of the COVID-19 vaccines, there has been an exponential growth in mRNA-based product development. These have moved from being almost exclusively vaccines to now encompassing many therapeutic applications ranging from gene editing and mRNA modified T cells to protein replacement. They span conditions including infectious diseases (e.g., RSV, influenza), cancer, and autoimmune diseases.
As mRNA products move through development, manufacturing changes will occur. These commonly occur to support requirements for late-phase clinical trials and, ultimately, commercialization. Typical changes include scale-up, changes made to improve product stability, formulation changes, changes to sources of critical materials and reagents, and manufacturing site changes (including adding new sites). As such, comparability is an essential component of the evolving manufacturing process.
A comparability exercise involves comparing the product before and after manufacturing changes and assessing the impact these changes may have on product quality attributes as they relate to safety and efficacy. It is important to consider where in the development life cycle the product is when designing a comparability plan. For products in early stages of development, analytical comparability may be sufficient.
Manufacturing changes made in later stages of development will require a more comprehensive study, and additional nonclinical and/or clinical studies may be needed to confirm the pre- and post-change material have comparable safety and efficacy. Therefore, manufacturing changes are not recommended during Phase 3 and/or registration studies.
Comparability Study Design
Planning for a comparability study should start with a description of the change(s) in the manufacturing process and the rationale for the change(s). Completing a risk assessment and designing the comparability study in such a way as to assess the potential impact of each change on product quality are key to a successful outcome.
In addition to having a good understanding of the manufacturing process (i.e., critical process parameters [CPPs]), product understanding, namely critical quality attributes (CQAs), and product characterization are also essential for comparability studies. It is important to identify what the critical attributes are and how they could be affected by the proposed manufacturing changes.
One item that is frequently overlooked in comparability study designs is the cumulative impact of individual changes. For example, when changing manufacturing sites, it is unusual for everything to stay the same. A new site may have different equipment and/or different materials. While individually these changes have minimal impact, when taken together there may be a significant impact on product quality, safety, or efficacy.
Execution of a comparability study requires a prospective study protocol with predefined acceptance criteria. When setting the acceptance criteria, consideration should be given to the criticality of the product attribute, sensitivity of the analytical assay, past manufacturing experience, and sources of variability. Data should be generated with both pre-change and post-change product to confirm compliance with product release specifications. Ideally, this should be a side-by-side analysis of multiple lots. Just because A=B and B=C, it does not necessarily mean that A=C. However, this approach may not be feasible when using patient-derived starting material. In that case, a split manufacturing approach can be employed (Figure 1). Irrespective of the approach taken, the study design should yield a statistically robust and comprehensive data set.
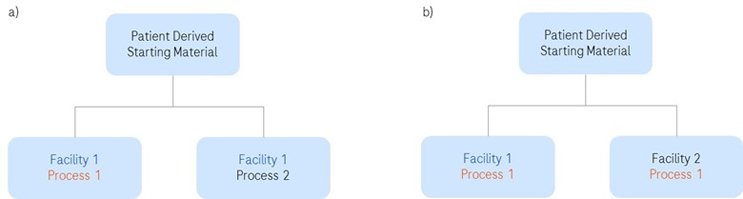
Figure 1: Examples of split manufacturing examples to demonstrate comparability from patient-specific starting material: a) new process implemented in one manufacturing facility; b) same process implemented into two manufacturing facilities.
Why Release Testing Is Just One Piece Of The Analytical Puzzle
Lot release testing in itself is not sufficient to evaluate the impact of manufacturing changes. An extensive data set should be generated evaluating critical stages in the process that could impact the final product characteristics, including in-process controls, drug substance release testing, drug product release testing, and extended characterization. It is also important to evaluate impurity profiles and to include stability data in the comparability exercise to identify differences in product degradation. For example, the manufacturing change may cause small changes in product-degrading enzymes that are not detectable by the characterization and product release testing.
Since the impact of subtle changes in product- and process-related impurities may only be detectable over a prolonged time, if possible, the stability study should include data from accelerated and stress conditions as well as real-time conditions.
The analytical methods used for assessment of comparability should be well-controlled and have sufficient accuracy, precision, specificity, and robustness to detect changes in the product. Where possible, assays should be validated. The analytical test panel should allow monitoring and control of process parameters as well as assessing the impact of changes on product CQAs.
The analytical methods used should include a biological potency assay and product characterization assays as the latter can identify changes to product attributes that are not examined with release testing. If possible, the use of orthogonal assays (i.e., more than one assay to evaluate a single CQA) can be beneficial. Perhaps the most important factor in determining the success of analytical comparability is reducing assay variability. This can be mitigated by testing pre- and post-change samples in the same assay run.
Another key consideration in the comparability exercise is the evaluation of historic data. This serves two main purposes: to set the study acceptance criteria and to provide insight into potential “drift” of quality attributes with respect to safety and efficacy. Development, nonclinical, engineering, and clinical batches should all be considered for inclusion in the manufacturing history evaluation.
Common Challenges For Comparability Of Cell And Gene Therapy Products
Cell and gene therapy products cover a very diverse set of modalities and product types, including, for example, mRNA-based therapies, DNA-based gene therapies, gene editing therapeutics (CRISPR/Cas9), viral vector-based therapies (e.g., AAV), and CAR-T therapies. However, there are common challenges when developing the comparability strategy for any CGT, including limited knowledge of CQAs, limited manufacturing experience, limited clinical experience, variable starting materials, small batch sizes, complex manufacturing processes, and short shelf life. Additionally, especially in early stages, analytical assays may not be fully developed (e.g., potency) and assays are unlikely to be qualified. There may be limited characterization data available and reference standards may yet to be established.
Manufacturers of cell and gene therapies, including mRNA products, are expected to comply with the current regulatory framework and guidance documents for conducting comparability. While the principles from ICH Q5E Comparability of Biotechnological or Biological Products Subject to Changes in Their Manufacturing Process1 are generally still applicable, these do not address the specific challenges of performing comparability studies with these types of products, and alternative approaches may need to be used.
For example, when using patient-specific starting materials it may not be possible to conduct side-by-side manufacture of pre-and post-change product. In this situation, the use of reference/retain samples from historic lots may be used for analytical comparability. Maintaining reference/retain samples may in itself be challenging due to factors including small batch sizes, storage conditions, and short shelf-life. Therefore, gaining as much data as possible from batches at the time of manufacture and release to leverage in future comparability evaluations may be the best option.
Ultimately, manufacturing processes for cell and gene therapies are complex, with a high degree of inherent variability; thus, focusing the comparability study on a specific stage of manufacturing may be the appropriate approach. Recognizing the need for alternative approaches, the U.S. FDA recently drafted comparability guidance for cell and gene therapy products2 and discussion of a comparability study design with the appropriate regulatory authorities is highly recommended.
Specific Considerations For mRNA-based Products
The scope of therapeutic uses for mRNA-based products spans a wide range and the scale and quality requirements for mRNA can be different and depend on the application. There will be varying demands in material, process, equipment, and testing at different scales. Therefore, it may be necessary to modify the standard comparability approach to account for the unique challenges of mRNA products.
The analytical test panel should still include in-process controls, drug substance release testing, drug product release testing, stability testing, and extended characterization. The differences are in the specifics needed to evaluate an mRNA product. For example, the characterization panel should include mRNA-specific attributes such as mRNA construct, plasmid sequence, and RNA modifications as well as detailed characterization of the delivery technology (e.g., lipid characterization for lipid nanoparticle [LNP] delivery). There should also be evaluation of functionality including transfection, expression, and functionality of the encoded sequence.
Scale Up Or Scale Out?
An interesting consideration for mRNA product development is whether to scale up or scale out the manufacturing process. There are two critical steps in mRNA manufacturing processes where scale-up is challenging. Scalable generation of mRNA molecules with high purity can be difficult as there is limited experience with development and characterization of the scalable purification methods. There are also scale-up limitations of HPLC-based purification processes, including the safety and environmental implications of using large volumes of solvents.
Perhaps the most significant scale-up challenge is the encapsulation step as the geometry of mixing setup and flow rates are quite specific. Even small changes in mixing geometry can change critical characteristics of the mRNA-LNP. Scaling up the manufacturing process would necessitate increasing the dimensions of the equipment that encapsulates the mRNA in the LNPs.
By instead scaling out — for example, replicating the process with more manufacturing units of the same size and design — the mixing geometry can be kept constant and thus mitigate any impact on LNP characteristics, which could affect efficacy and safety.
Scaling out a process would not be expected to impact product quality attributes and, therefore, a comparability study would be unnecessary. However, scaling out frequently involves moving the process to a larger and/or additional manufacturing site, in which case a comparability study is required.
Final Thoughts
Comparability is one of the most common CMC concerns regulatory agencies have at late stages of product development.3 Therefore, well designed and executed comparability studies are essential as mRNA products advance through the development life cycle.
Comparability studies should start with quality data and then continue as appropriate with non-clinical and clinical studies. The extent of the studies needed will depend on where in the manufacturing process the changes are being made, the potential impact on product quality attributes, safety and efficacy, and the suitability of analytical techniques to detect potential product modifications. The unique challenges developing mRNA products add complexity to the standard approach to comparability. However, these can be mitigated with careful planning, thus leading to the successful development of these products.
References
- ICH Guidance: Q5E Comparability of Biotechnological or Biological Products Subject to Changes in Their Manufacturing Process
- FDA Draft Guidance for Industry: Manufacturing Changes and Comparability for Human Cellular and Gene Therapy Products July 2023
- Barkholt, L., Voltz-Girolt, C., Raine, J. et al. European regulatory experience with advanced therapy medicinal products. Nat Rev Drug Discov 18, 8–9 (2019).
About The Author:
 Elaine Peters, Ph.D., is a director of analytical capabilities in cell and gene therapy technical development at Genentech. She is responsible for the strategic framework establishing analytical and QC capabilities to support the commercialization of the cell and gene therapy portfolio. She has expertise in analytical development and quality control. She holds a Ph.D. in neuroscience from the University of Glasgow in Scotland and an executive certificate in leadership and management from The Wharton School at the University of Pennsylvania.
Elaine Peters, Ph.D., is a director of analytical capabilities in cell and gene therapy technical development at Genentech. She is responsible for the strategic framework establishing analytical and QC capabilities to support the commercialization of the cell and gene therapy portfolio. She has expertise in analytical development and quality control. She holds a Ph.D. in neuroscience from the University of Glasgow in Scotland and an executive certificate in leadership and management from The Wharton School at the University of Pennsylvania.