
Strategies To Tackle CAR-T Product Challenges
By Peter H. Calcott, Ph.D., FRSC, president and CEO, Calcott Consulting LLC

Since 2017, at least six CAR T cell therapy products have been licensed in the U.S.1 While this is a major advance in the treatment of cancers, it has come at a very high treatment price versus classical biopharmaceuticals, which has created quite a stir in the market place.2 These products and other cell and gene therapy products all share common elements associated with their manufacture. All of them are examples of personalized medicines where the batch that is released is designated for a single patient versus an exceedingly large number of patients, as is for classic drugs, including biopharmaceuticals. Because of this, it has been difficult to scale up operations and take advantage of the reduced cost associated with large-scale operations. In this article I will describe the challenges that these types of medicines face and how pharma has attempted to combat these scale-up and other challenges.
Setting Your Sights On The Challenges
Classical pharmaceuticals in the clinical phase are manufactured in small batches and are inherently expensive to produce, including testing. But once through clinical trials and into the commercial world, scale-up can reduce the cost per dose considerably. The fixed costs (e.g., batch testing) can now be spread across thousands of doses versus tens to hundreds or even less. While a clinical batch may be small (100 to 1,000 doses), commercial operations yield batches of 10,000 to 100,000 doses.
Typical cell and gene therapy products (autologous) are equally expensive or even more so at the early clinical phase, but as they are commercialized, we are not seeing the advantages of scale that have been evident in more classical biopharmaceuticals. Why is that? It is because there is no obvious way to scale up to take advantage of the economies of scale. We saw somewhat the same issue in the 1980s when some of the first biopharmaceuticals were commercialized. Some companies such as Genentech moved from small-scale cell culture vessels (20 L to 100 L) to increasingly larger vessels of 10,000 L to 20,000 L. Other companies such as Amgen, which had invested in roller bottle technology, scaled up by adding more roller bottles. This is precisely the same challenge we see today in cell and gene therapy.
Therefore, to take advantage of the efficiencies of scale, companies will need to make a radical shift in operations. These approaches fall into several types. But to understand these we must appreciate the pain points in the technology. Although each of the products listed (and others venturing through pipelines) is unique, they do share the same common approach, which is shown in Figure 1.
Basically, cells are harvested from the patient via processes such as apheresis and then are treated with the therapy agent, after which the activated cells are expanded and subsequently harvested, where they can be cryopreserved, if necessary. At this point, the material is assessed for acceptability by quality assurance, including any QC testing required. This constitutes the batch at this stage. As appropriate, the product is reinfused into the patient. In most products, the apheresis to harvest the patient’s cells is performed at the bedside while the activation, expansion, and harvesting are performed in the “factory” (Figure 1). After this, the cells are returned to the patient for infusion. Therefore, the cells are transported twice, which is a logistical and technical challenge. This point is pertinent in further discussion on optimization.
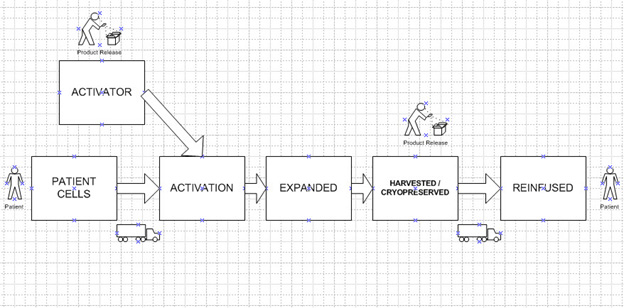
Figure 1: Schematic Process for CAR-T Therapy. Note there are two transportations and most manufacturing occurs at the factory. One batch is produced per patient. The activator is used for multiple patients.
Unlike the final product (infused into patients), there is also a second product to consider and that is the activating agent. This agent (often a gene cloned into a viral vector) is product specific but can be manufactured in large batches to treat hundreds or thousands of patients. It can be scaled up easily and thus we can gain efficiency from its scale.
(Some) Improvements
Incremental improvements in efficiency of scale have been made with a variety of technologies, including automation and robotics to decrease manpower requirements. Other improvements have been in the so called “All in One” operations,3,4 suite design, and the “process in a box” concept. Basically, all operations are performed within an isolator to reduce the risk of open operations versus outside in laminar flow hoods. But as indicated, these are just incremental improvements. They do not even approach the efficiencies of large scale seen before in classical biopharmaceuticals.
The strategies employed to make a quantum shift in efficiency fall into two major types: operational optimization and innovation of process strategy.
Operational Optimization
Moving Manufacturing Away From The Factory Toward The Bedside
This novel approach is to eliminate the steps that are performed in the factory (cell activation, proliferation, harvesting, storage, etc.) and eliminate the transportation steps and perform these process steps at the bedside or, at least, in the patient’s medical facility. This is not a new concept. In fact, even with a lyophilized product, the reconstitution with WFI and dissolution is routinely performed at the bedside. This step is not considered a manufacturing step but rather a routine operation. The drug company performs many studies to define all critical processes involved, eliminating risk to the patient, including advising on aseptic processes and stability of the reconstitution of the solution. These are included in the submission to the agencies and are part of the product review. At-home Covid test kits also use elements of this principle, with the patient performing the test at home. But for it to be successful, it must be foolproof. With cell therapy, we are not quite there yet.
The Hub Model
Companies have not been able to eliminate the manufacturing steps completely and relocate them all to the bedside, but progress has been made with the hub model. That is the establishment of “centers of excellence” in key hospitals, where these crucial steps are performed. The final product is still released through conventional GMP processes at the bedside. However, this model means that there will be a finite number of locations that can perform these treatments, as Figure 2 illustrates. This model greatly reduces the need for transportation of patients’ cells and the inherent risks and logistics challenges. But it requires extensive training and oversight at the hubs. It still does not allow for the efficiency of scale to be captured because the product is still patient specific.
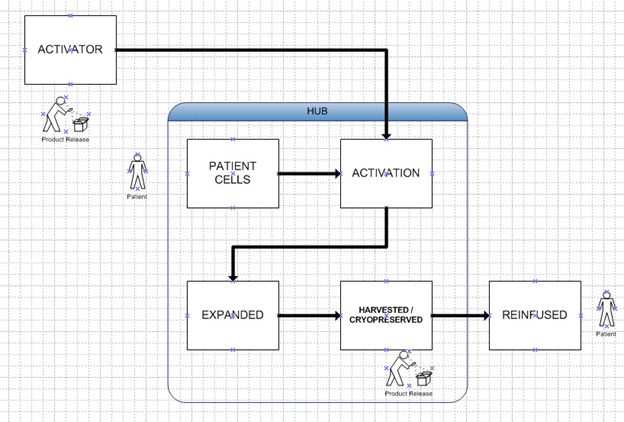
Figure 2: Hub Model. Same as Figure 1 except all manufacture of activated cells takes place at a hospital center. It still produces one batch per patient.
The Bedside Model
The next step might be the “fully distributed model” or bedside model, where treatment can be done at essentially any competent medical facility. This may eventually occur, but not in the immediate future. With this model the product released is not the cells that have been activated but instead it is the activating agent and any other elements of the treatment. That means it can easily be scaled up and take advantage of the efficiencies of scale. Note that these batches now can treat hundreds if not thousands of patients. This is illustrated in Figure 3.
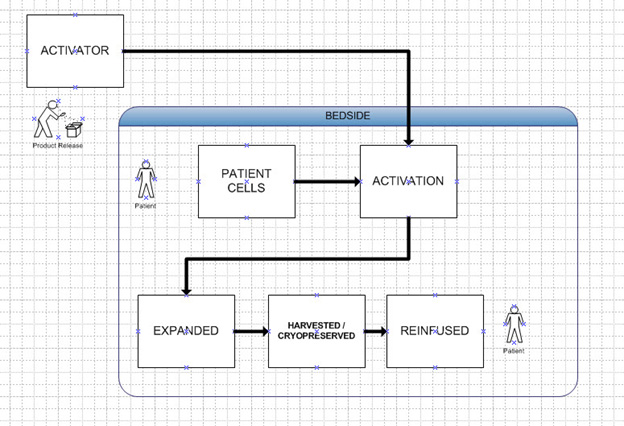
Figure 3: Bedside Model. Same as Hub, except activity at hospital is not considered manufacturing like vial reconstitution. Activator batch can supply multiple patients.
Innovation Of Process Strategy
Using the patient’s cells as the basis of treatment using the present strategies will always lead to the individual batch being specific to each patient unless some radical changes are made to simplify operations. Companies appear to be pursuing two strategies.
In Vivo Proliferation
In this approach, we eliminate the cell collection, treatment, proliferation, harvesting, and reinfusion steps and instead perform these steps in vivo5. Thus, the treatment uses the activator therapy (gene cloned in vector), which is injected together with elements that will allow the agent to be absorbed by relevant cells and establish itself, allowing cells to proliferate in the body. The nature of these agents might include nanoparticles, exosomes, or other technologies. Thus, the product is a multi-patient product comprising the activator and proliferation elements. This can then be easily scaled up. Again, the advantage of this is that a batch of material can be used to treat hundreds or thousands of patients because it is not patient specific. This is illustrated in Figure 4.
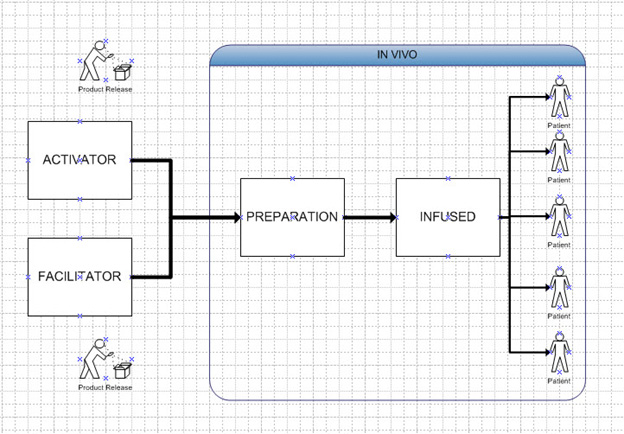
Figure 4: In Vivo Model. Activator and facilitator are the products. One batch of activator and facilitator can supply multiple patients.
Using Pluripotent Or Stem Cells
This option uses the allogenic cell approach. That is, it uses pluripotent or stem-like cells that can be transformed in the laboratory, proliferated, harvested, stored, and infused in patients. Thus, the product is like the classical CAR T product, except it is not using the patient’s own cells but rather cells that are “generic,” usable by a wide range of patients. Discgenics has performed some ground-breaking work in this area in spine disc regeneration.6 The problems of QC testing (see the next challenge) and scale-up are eliminated. Again, a batch of product can treat hundreds or thousands of patients. Figure 5 illustrates this.
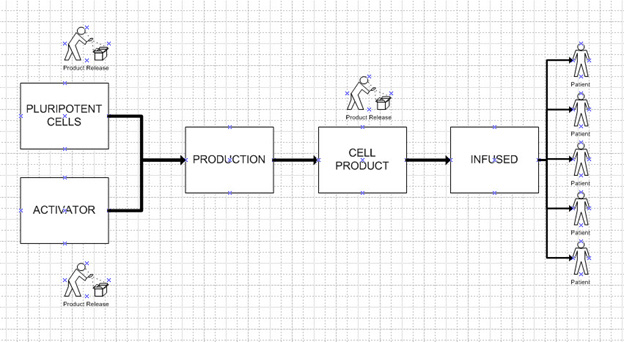
Figure 5: Pluripotent Cell Model. One batch of cell product can supply multiple patients. Most operations are performed at the factory.
Comparison Of All Models
Table 1 illustrates all these models and compares their attributes. The key to the table is that the first three models (present, bedside release, and hub) all rely on the patient’s cells as the final release. Thus, each batch can only treat a single patient. On the other hand, the last three models (bedside treatment, in vivo, and pluripotent models) all rely on the final release being the activating agent, which is scalable and able to treat a large number of patients per batch. We can do that because we no longer are releasing the patient’s cells in the bedside treatment and the in vivo model. We have established a process of cell activation that is no longer considered manufacturing but rather is like reconstitution of the lyophilized product – a simple process. That is, it is foolproof and able to be performed reliably and reproducibly without imposition of GMP standards. The pluripotent model still requires activated cells to be released, but these can be used to treat hundreds of patients per batch and hence can be readily scaled to benefit from the efficiencies of scale.
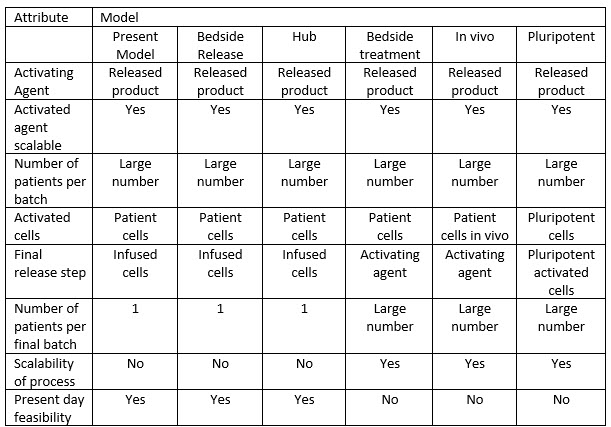
Table 1: Comparison of all types
Quality Control Challenges To Keep In Mind As You Move Forward
Besides the challenges to conventional scale-up, as outlined above, there are challenges in production strategies. In many cases, the activated cells must be reinfused into patients rapidly, which poses several QC issues. While many of the tests for release can be turned around in one day, several of the tests are more time-consuming. Because many of the operations described are considered open, contamination of product is a significant risk. Conventional biological tests such as sterility, mycoplasma, or general virus testing take much more than a day, upward of 14 to 30 days to complete. While there have been advances in rapid testing for sterility, mycoplasma, and specific viruses that are gaining traction, none to date can be directly substituted for the longer tests. So, it is not uncommon for companies to develop other strategies for more rapid release of product. This includes relying on risk management, the rapid surrogate tests, and peripheral testing (environmental monitoring aseptic process simulations, etc.) to assure sterility rather than the classical longer tests described. In many cases, products are released based on testing that can be completed in the allotted time and then reporting the sterility test post product usage (with concurrence from regulatory bodies). In these cases, the sterility assurance program is paramount. The use of isolator and robotic technologies has strengthened these strategies. But there is always the potential of a product being administered to a patient, meeting all release criteria at that time, only to have it fail a sterility test after administration. If these cells could be frozen until conventional testing is completed, that would alleviate these challenges. At present the most common cryoprotectant is DMSO but it poses toxicity challenges. Recently a novel cryoprotectant, peptoids, have been developed by X-Therma.7 Their cryoprotectant XT Thrive is a protein-free, DMSO-free cryoprotectant. They have recently submitted a DMF to the FDA.
There is also the potential of a patient’s cells failing the release tests prior to treatment. This leaves the company and physician in a dilemma: whether to use the product, which may be life saving, or not releasing it. This article focuses on the technical issues of these types of products and not the ethics of these challenges.
These strategies help to overcome the urgency of lot disposition, but they do not alleviate the economies of scale issues discussed above.
Conclusions
The field of cell and gene therapy, often referred to as regenerative medicine, is rapidly advancing. The challenges discussed here are partly solved, and continual progress will be made in the areas of efficacy, safety, and affordability of treatments in the future.
References
- Kymriah – Novartis (2017), Yescarta – Kite (2017), Tecartus – Kite (2020), Breyanzi – BMS (2021), Abecam – BMS (2021), Carvykti – Janssen (2022)
- Edward R. Scheffer Cliff et al. (2023) High Cost of Chimeric Antigen Receptor T-Cells: Challenges and Solutions American Society of Clinical Oncology Educational Book. Volume 43. https://doi.org/10.1200/EDBK_397912
- Alzubi, Jamal, Dominik Lock, Manuel Rhiel, Sabrina Schmitz, Stefan Wild, Claudio Mussolino, Markus Hildenbeutel et al. "Automated generation of gene-edited CAR T cells at clinical scale." Molecular Therapy-Methods & Clinical Development 20 (2021): 379-388.
- O'Connor, Joseph, Kalyani Daita, Janet Sei, Eytan Abraham, and Yaling Shi. "Manufacturing of patient specific novel T cell therapies using the Cocoon Platform automated system." (2022).
- Stanton, D. (2023). Novartis at JPM: ‘7-day CAR-T process an alternative to allogeneic.’ In Bioprocess International.
- Engineering Discogenic Cells https://www.discgenics.com/research-and-development
- X-Therma develops a DMSO-free, protein-free cryoprotectant. www.x-therma.com
About The Author:
 Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.
Peter H. Calcott, D.Phil., is president and CEO of Calcott Consulting LLC, which delivers solutions to pharmaceutical and biotechnology companies in the areas of corporate strategy, supply chain, quality, clinical development, regulatory affairs, corporate compliance, and enterprise e-solutions. He has also served as an expert witness. He also teaches at the University of California, Berkeley in the biotechnology and pharmaceutics postgraduate programs. Previously, he was executive VP at PDL BioPharma, chief quality officer at Chiron and Immunex Corporations, and director of quality assurance for SmithKline Beecham and for Bayer. He has also held positions in R&D, regulatory affairs, process development, and manufacturing at other major pharmaceutical companies. He has successfully licensed products in the biologics, drugs, and device sectors on all six continents. Calcott holds a doctorate in microbial physiology and biochemistry from the University of Sussex in England. He has been a consultant for more than 20 years to government, industry, and academia.